




用户文章 IF=19.2|范先群院士团队利用m1A MeRIP-seq联合多组学测序揭示眼黑色素瘤调控新机制
发布时间:2024-01-17 13:47 | 点击次数:
N1-甲基腺苷(m1A)是一种普遍、可逆的转录后RNA修饰,可修饰tRNA、rRNA和mRNA。m1A RNA修饰的精确时空调控对于表观基因组稳态至关重要,可影响RNA的二级结构和蛋白质-RNA相互作用。并且,目前大量新的研究表明,动态的m1A甲基化在基因表达、RNA稳定性和翻译效率等生物学过程中也起着至关重要的作用。其中值得我们注意的是,m1A甲基化还参与多种癌症的癌变,包括胃肠道癌、口腔鳞状细胞癌、宫颈癌和淋巴瘤等。异常的m1A甲基化能够引发一些致癌事件,比如代谢重编程、胶原蛋白产生以及tRNA衍生出小RNA。
虽然在许多癌症研究中经常观察到m1A去甲基化酶ALKBH3的表达增加,能加速恶性增殖和侵袭。但是,我们对于这些m1A甲基化修饰在致癌中的潜在功能却仍然知之甚少!
2023年12月1日,云序客户上海交通大学医学院附属第九人民医院范先群院士团队在Nucleic Acids Research(IF=19.16)上发表了题为“Histone lactylation-boosted ALKBH3 potentiates tumor progression and diminished promyelocytic leukemia protein nuclear condensates by m1A demethylation of SP100A”的研究性论文。该研究通过ChIP-seq、CUT&Tag、m1A MeRIP-seq、RNA-seq、LC-MS等多组学联合分析,揭示了在眼部黑色素瘤中组蛋白乳酸化能够导致ALKBH3表达增加,m1A修饰特异性降低,进而降低急性早幼粒细胞白血病蛋白(PML)聚集并促进肿瘤恶化。总体而言,研究结果表明依赖ALKBH3的m1A去甲基化对于PML核体的形成至关重要,提供了一种新的治疗策略,其中“靶向m1A重编程策略”对肿瘤治疗有效。
该研究中的m1A MeRIP-seq和RNA-seq高通量测序技术均由上海云序生物公司提供。这是云序生物与该课题组继m6A MeRIP-Seq 合作后,再次发表的RNA甲基化高分文章。

标题:Histone lactylation-boosted ALKBH3 potentiates tumor progression and diminished promyelocytic leukemia protein nuclear condensates by m1A demethylation of SP100A
发表时间:2023年12月1日
发表期刊:Nucleic Acids Research
影响因子:IF=19.16/Q1
研究方法:ChIP-seq、CUT&Tag、蛋白质组学分析(LC-MS)、m1A MeRIP-seq、 RNA-seq、RIP-qPCR、荧光素酶报告实验、 多聚核糖体分析(Polysome profiling)
研究摘要
尽管m1A RNA修饰是RNA代谢的重要调节因子,但m1A修饰在癌变中的作用仍然是谜。本研究发现,组蛋白乳酸化通过去除SP100A的m1A甲基化,增强了ALKBH3的表达,同时减弱了肿瘤抑制性急性早幼粒细胞白血病蛋白(PML)凝聚物的形成,促进癌症的恶性转化。首先,由于组蛋白乳酸化水平过高,ALKBH3在高危眼黑色素瘤中特异性上调,导致m1A低甲基化状态。随后得多组学分析确定了PML小体的核心成分SP100A是ALKBH3的下游候选靶点。在治疗上,沉默ALKBH3在体外和体内对黑色素瘤都显示出有效的治疗效果,这可以通过耗尽SP100A来逆转。在机制上,我们发现YTHDF1负责识别m1A甲基化的SP100A转录物,能够增加其RNA稳定性和翻译效率。最后,我们初步证明了m1A修饰是肿瘤抑制基因表达所必需的,扩大了目前对肿瘤进展过程中m1A动态功能的理解。此外,我们的研究结果表明,乳酸化驱动的ALKBH3对于PML核凝聚物的形成是必不可少的,它连接了我们对m1A修饰、代谢重编程和相分离事件的了解。

图文摘要
技术路线

*红色部分技术服务云序均可提供
研究结果
(1)ALKBH3在眼黑色素瘤中特异性升高,并与不良预后相关
为了确定m1A修饰在眼部黑色素瘤发病机制中的作用,我们首先比较了眼黑色素瘤样本和对照样本的整体m1A修饰水平。值得注意的是,通过Dot blot实验证实了眼黑色素瘤样本的m1A水平显著降低(图1A-B)。此外,通过WB和qPCR等实验证实了去甲基化酶ALKBH3在肿瘤中的蛋白表达增加(图1C-E),并且ALKBH3升高与不良结果相关(图1F),进一步强调了ALKBH3在眼黑色素瘤癌变中的重要性。
与正常色素细胞(PIG1)相比,大多数黑色素瘤细胞系(MUM2B、OCM1、OMM2.3、OMM1、MEL290、92.1、CRMM1、CRMM2、CM2005.1)中m1A水平显著降低(图1G-H), ALKBH3水平显著升高(图1I-K)。此外,RNA-seq进一步证实ALKBH3在眼黑色素瘤细胞系中表达上调(图1L)。并且在这些细胞中,ALKBH3与m1A水平呈显著负相关,表明了ALKBH3上调导致了m1A水平降低(图1M)。
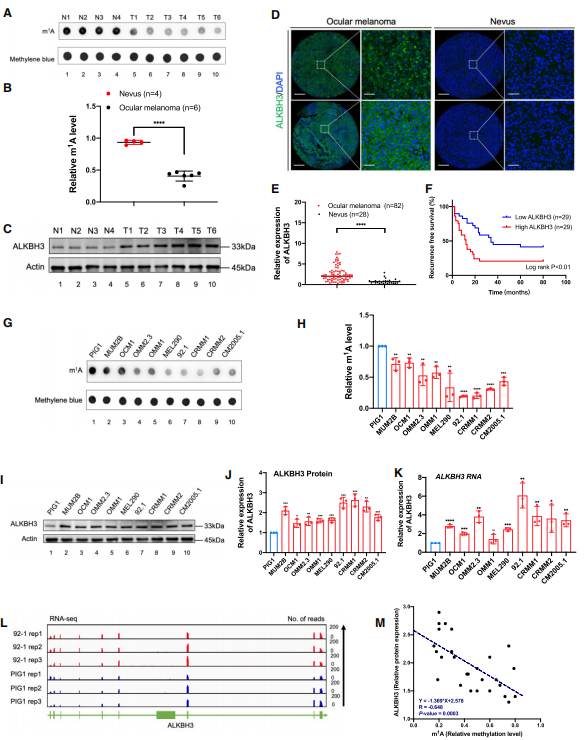
图1:眼黑色素瘤中ALKBH3表达增加和m1A水平降低,且高ALKBH3与较低生存率相关
(A)Dot blot显示肿瘤和正常样本中相对于亚甲基蓝信号的m1A信号。
(B)密度分析显示肿瘤和正常样本中m1A相对于亚甲基蓝的表达。
(C) Western blot显示肿瘤和正常样本中ALKBH3相对于Actin的表达。
(D)肿瘤和正常样本中ALKBH3(绿色)和DAPI(蓝色)的免疫荧光。
(E)正常组织和肿瘤组织中ALKBH3水平的统计结果。
(F)低ALKBH3水平(n = 29)和高ALKBH3水平(n = 29)眼黑色素瘤患者肿瘤复发Kaplan-Meier曲线的差异。
(G)Dot blot显示眼黑色素瘤细胞和正常黑色素细胞中相对于亚甲基蓝信号的m1A信号。
(H)密度分析显示m1A相对于亚甲基蓝在眼黑色素瘤细胞和正常黑色素细胞中的表达。
(I) WB显示ALKBH3相对于ACTB在眼黑色素瘤细胞和正常黑色素细胞中的表达。
(J)密度分析显示ALKBH3蛋白相对于ACTB蛋白在眼黑色素瘤细胞和正常黑色素细胞中的表达。
(K) qPCR数据显示ALKBH3在眼黑色素瘤细胞中的表达相对于PIG1细胞。
(L) IGV从眼黑色素瘤细胞系(92.1)和正常黑色素细胞细胞系(PIG1)的RNA-seq数据中追踪ALKBH3。
(M)眼黑色素瘤细胞和正常黑色素细胞中ALKBH3蛋白相对表达与m1A甲基化水平的相关性分析。
(2)ALKBH3在体外和体内加速眼黑色素瘤的形成
为了探索ALKBH3的致癌功能,使用两个单独的shRNA沉默了ALKBH3的表达(图2A-B)。同样,在ALKBH3缺失的细胞中观察到m1A水平显著增加(图2C)。此外,ALKBH3沉默导致所有眼黑色素瘤细胞的细胞生长和聚集能力显著衰减(图2D-F)。Transwell实验显示,ALKBH3的敲低导致迁移能力下降(图2G和H)。这些数据支持ALKBH3是体外眼黑色素瘤恶性增殖和转移的必要致癌因子。为了评估它们在体内的肿瘤形成能力,我们将对照和ALKBH3沉默的92.1黑色素瘤细胞(荧光素酶标记)注射到裸鼠体内,并在原位异种移植模型中监测肿瘤生长。生物发光成像显示,缺乏ALKBH3的眼黑色素瘤细胞的信号强度比对照细胞弱(图2I)。此外,在ALKBH3沉默组中,异种移植物的平均重量减少了约80%(图2J)。综上所述,这些实验表明,ALKBH3在体外和体内的眼黑色素瘤的发生中起着致瘤作用。
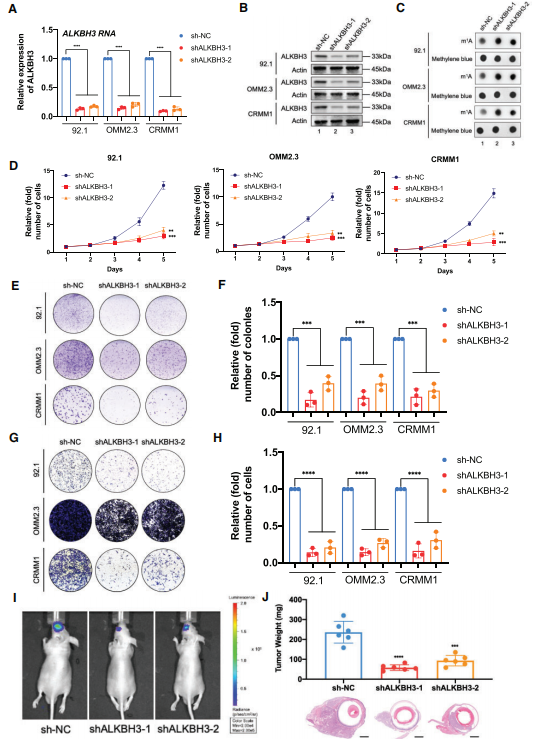
图2:ALKBH3敲低可增加m1A水平,抑制眼部黑色素瘤的发生
(A) qPCR数据显示ALKBH3敲低后在眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)中表达ALKBH3。
(B) Western blot显示ALKBH3敲低后,眼部黑色素瘤细胞(92.1、OMM2.3和CRMM1)中ALKBH3相对于ACTB的表达。
(C)Dot blot显示ALKBH3敲低后眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)中m1A信号相对于亚甲基蓝信号的变化。
(D)采用CCK-8法评估ALKBH3敲除后眼部黑色素瘤细胞(92.1、OMM2.3和CRMM1)的增殖情况。
(E)采用集落形成试验评估ALKBH3敲除后眼部黑色素瘤细胞(92.1、OMM2.3和CRMM1)的生长情况。
(F) ALKBH3敲低后眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)集落形成实验数据的统计分析。
(G)采用transwell实验评估ALKBH3敲除后眼部黑色素瘤细胞(92.1、OMM2.3和CRMM1)的迁移情况。
(H) ALKBH3敲低后眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)的transwell检测数据统计分析。
(I)体内小动物成像系统获得的图像显示,来自ALKBH3缺陷92.1细胞的原位异种移植物中生物发光信号受到抑制。
(J)缺乏ALKBH3的92.1细胞的同种异种移植物的重量直方图。H&E染色显示肿瘤组织。
(3)组蛋白乳酸化增强了ALKBH3的过度表达
为了确定ALKBH3表达增加的分子基础,我们查询TCGA数据库,筛选与ALKBH3具有平行表达模式的基因。通过GO和KEGG分析,我们发现ALKBH3相关基因在氧化磷酸化、细胞代谢过程和碳水化合物衍生物代谢过程等代谢过程中富集。表明ALKBH3 RNA表达的升高可能与代谢重编程有关。此外,乳酸生成酶LDHA和LDHB与ALKBH3呈显著正相关,这表明ALKBH3与乳酸之间存在关联(图3A-B)。由于先前的研究表明组蛋白乳酸化有助于癌基因的激活,并且眼黑色素瘤的乳酸化水平升高,我们认为ALKBH3水平的升高可能与组蛋白乳酸化有关。
然后,我们验证了泛乳酸化和组蛋白乳酸化标记物(H3K18la)在眼部黑色素瘤队列中的表达模式,显示与ALKBH3蛋白表达显著正相关(图3B-C)。更重要的是,H3K18la分析的CUT&Tag和ChIP-Seq都显示了在ALKBH3启动子区域捕获的组蛋白乳酸化信号的强大信号。此外,组蛋白乳酸化抑制剂(草酸酯和2-DG)和LDHA/B抑制均导致ALKBH3启动子区域组蛋白乳酸化水平显著降低(图3F),随后,这两种抑制均导致ALKBH3启动子区域组蛋白乳酸化水平显著降低在所有测试的黑色素瘤细胞中去除ALKBH3的RNA和蛋白质水平(图3G-J)。

图3:组蛋白乳酸化增强了ALKBH3的表达
(A) TCGA中ALKBH3表达与LDHA或LDHB表达的相关性分析(n = 80)。
(B)肿瘤和正常标本中ALKBH3(绿色)、Pan Kla(红色)和DAPI(蓝色)的免疫荧光。ALKBH3相对蛋白表达与Pan Kla水平在眼黑色素瘤细胞和正常黑色素细胞中的相关性分析。
(C)肿瘤和正常样本中ALKBH3(绿色)、H3K18la(红色)和DAPI(蓝色)的免疫荧光。眼部黑色素瘤细胞与正常黑色素细胞中ALKBH3、H3K18la相对蛋白表达水平的相关性分析。
(D) ChIP-seq分析的IGV轨迹显示ALKBH3启动子处H3K18la富集。
(E)来自CUT&Tag分析的IGV轨迹显示在ALKBH3启动子处H3K18la富集。
(F)用组蛋白乳酸化抑制剂(2-DG或草酸酯)或LDHA/B抑制治疗后,眼黑色素瘤细胞(92.1和CRMM1) ALKBH3基因组区域H3K18la状态的ChIP-qPCR检测。
(G) qPCR数据显示,在组蛋白乳酸化抑制剂(2-DG或草酸酯)或LDHA/B抑制治疗后,ALKBH3在眼黑色素瘤细胞(92.1和CRMM1)中表达。
(H和I) Western blot显示,在组蛋白乳酸化抑制剂(2-DG或草酸酯)作用下,眼黑色素瘤细胞(92.1和CRMM1)中ALKBH3相对于ACTB和Pan Kla的表达,H3K18la相对于Histone H3的表达。
(J) Western blot显示LDHA/B抑制后眼黑色素瘤细胞(92.1和CRMM1)中LDHA、LDHB、ALKBH3相对于ACTB的表达和Pan Kla、H3K18la相对于组蛋白H3的表达。
(4)多组学筛选发现SP100A是ALKBH3的下游候选基因
然后,作者探讨了ALKBH3沉默对眼部黑色素瘤细胞的抑制作用的机制。由于ALKBH3负责去除m1A修饰,首先对眼黑色素瘤细胞和正常黑素细胞进行了m1A MeRIP-seq。结果表明,从正常细胞和肿瘤细胞生成m1A MeRIP-seq文库中,平均鉴定出16 864个和10 212个m1A峰(图4A-B)。与之前的m1A MeRIP-seq结果一致,m1A峰在5个UTR中富集,特别是在起始密码子附近。差异表达的m1A修饰基因与多种黑色素瘤相关通路相关,包括DNA复制、mTOR/AMPK信号通路和黑色素合成,提示m1A修饰在眼黑色素瘤的发病机制中具有调控作用。
此外,在沉默ALKBH3后,作者进行了一系列全面的高通量筛选,包括m1A MeRIP-seq、RNA-seq和黑色素瘤细胞系的蛋白质组学分析(iTRAQ,图4F)。同样,我们注意到m1A修饰位点的变化在肿瘤相关通路中显著富集,包括PI3KAkt信号通路、局灶粘附和蛋白聚糖合成(图4C)。此外,ALKBH3的沉默导致了基因表达水平的显著变化,其中有199个上调基因,74个下调基因。同样,观察到蛋白质组水平的显著变化(149个上调蛋白,67个下调蛋白,图4F),进一步强调了ALKBH3在眼黑色素瘤发病机制中的重要性。
结合这些多组学数据,我们注意到在沉默ALKBH3后,眼黑色素瘤细胞中的核自身抗原斑点蛋白100 (SP100)在mRNA和蛋白水平上都上调,随后m1A修饰水平发生了巨大变化(图4G-M)。值得注意的是,SP100负责PML核小体的形成,主要作为各种癌症类型的肿瘤发生抑制因子,包括黑色素瘤、胶质母细胞瘤、平滑肌肉瘤、乳腺癌和喉癌。这一观察结果与在ALKBH3缺陷细胞中观察到的抑制效果一致。与正常黑色素细胞相比,SP100显示眼黑色素瘤细胞中的m1A甲基化降低。m1A MeRIP-seq和m1A-MeRIP-qPCR都证明了ALKBH3抑制后,眼黑色素瘤细胞中m1A修饰水平的下降得到了恢复(图4J)。由于最近的研究发现mRNA的m1A修饰增强了基因的表达和翻译效率,因此可能ALKBH3介导的m1A修饰对肿瘤抑制因子SP100的表达至关重要。然后,我们通过RNA-seq和qPCR验证了抑制ALKBH3后SP100在mRNA水平上的显著上调(图4K-L)。iTRAQ和Western blot检测显示,在ALKBH3抑制的细胞中,SP100蛋白水平也同样升高(图4M)。同样,ALKBH3在87例转移性黑色素瘤样本中与SP100呈显著负相关。综上所述,这些数据表明ALKBH3可能通过去除其m1A修饰来抑制SP100的表达水平。

图4:ALKBH3通过去除m100修饰来抑制SP100的表达
(A) m1A-MeRIP-seq数据显示了在眼部黑色素瘤细胞和正常黑素细胞中发现的m1A峰内的顶部富集基序。
(B) Pie图显示了眼部黑色素瘤细胞和正常黑色素细胞中不同RNA区域(CDS、5’UTR、3’UTR、起始密码子和终止密码子)的m1A峰分布。
(C)野生型和ALKBH3缺失的眼部黑色素瘤细胞中m1A修饰基因的KEGG通路分析。
(D)火山图显示了野生型和ALKBH3缺陷的眼部黑色素瘤细胞中ALKBH3调控的基因。
(E)野生型和ALKBH3缺陷的眼部黑色素瘤细胞中ALKBH3调控基因的KEGG通路分析。
(F)条形图显示了ALKBH3调控基因在宽型和ALKBH3缺失的眼黑色素瘤细胞中的蛋白表达。
(G-H)多组学分析确定SP100为ALKBH3的下游靶点。
(I)来自m1A-MeRIP-Seq分析的IGV轨迹显示m1A在SP100A的5UTR处富集。
(J) m1A-MeRIP- qPCR检测野生型和ALKBH3缺陷眼黑色素瘤细胞(92.1、OMM2.3和CRMM1) SP100中m1A的状态。
(K)从野生型和ALKBH3缺陷眼黑色素瘤细胞的RNA-seq数据中,IGV追踪SP100。
(L) qPCR数据显示,ALKBH3敲低后,眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)中SP100 RNA表达。
(M) Western blot显示野生型和ALKBH3缺陷眼黑色素瘤细胞(92.1,OMM2.3和CRMM1)中SP100相对于ACTB的表达
(5)SP100A在眼部黑色素瘤中作为肿瘤抑制因子
在病毒感染反应的背景下,SP100的四种主要亚型(即SP100A、SP100B、SP100C和SP100HMG)已经被识别出来,这促使我们对这些转录本所表现出的表达水平进行比较分析。值得一提的是,SP100A与SP100B、SP100C和SP100HMG在前15个外显子(1 - 1562 bp)上具有共性,而最后一个外显子的长度差异为395 bp。从眼黑色素瘤细胞(92.1)和正常黑色素细胞(PIG1)中获得的RNA-seq数据显示SP100A中有一个突出的信号,而SP100A不包括的外显子中的信号可以忽略不计(图5A)。此外,我们使用各种引物进行qPCR。这些引物包括一组专门为SP100A设计的引物(称为SP100-P1),以及另一组检测SP100B、SP100C和SP100HMG的引物(称为SP100-P2)。在眼黑色素瘤细胞和正常色素细胞中观察到SP100A的RNA表达(图5B)。由于有报道称人乳腺癌细胞系ZR-75-1表达SP100HMG,我们将ZR-75-1细胞作为阳性对照。重要的是,SP100B/SP100C/SP100HMG仅在ZR-75-1中检测到,而在其他细胞系中未检测到。此外,四种同工异构体的分子量也存在差异。Western blot结果显示,SP100A蛋白在所有被测细胞系中特异性表达(~ 72 kDa) ,这与之前研究中SP100A的分子量一致。研究结果表明,SP100A在我们的实验环境中大量表达,而其他亚型(SP100B、SP100C和SP100HMG)的表达可以忽略不计。
通过RNA-seq、qPCR和Western blot检测,发现与正常色素细胞相比,在眼黑色素瘤细胞中也观察到SP100A的RNA表达水平显著降低(图5A-C)。为了充分揭示SP100A在眼部黑色素瘤中的功能,我们检测了SP100A在眼部黑色素瘤临床样本中的表达。我们发现SP100A在眼部黑色素瘤样本中明显降低(图5D和E)。更重要的是,在我们的队列和TCGA队列(图5G)中,SP100A的缺失都与不利的结果相关(图5F-G)。
此外,根据眼部黑色素瘤样本的单细胞分析,SP100的表达与癌症激活标志评分降低有关,包括侵袭、转移、细胞周期激活、增殖和上皮-间质转化。总的来说,这些数据表明SP100A在眼部黑色素瘤中下调并可能抑制几种致癌事件。
由于SP100A在眼黑色素瘤中下调,我们在三种眼黑色素瘤细胞系中外源性过表达SP100A。有趣的是,所有测试的眼部黑色素瘤细胞在过表达SP100A后,增殖能力都减弱(图5H)。此外,与对照组相比,过表达SP100A的黑色素瘤细胞形成的菌落更小、更少(图5I和J)。此外,在眼部黑色素瘤细胞中引入SP100A后,观察到肿瘤转移能力明显受到抑制(图5K和L)。
由于SP100A在PML小体中充当分子支架,我们随后评估了其在PML中的作用,PML在过表达SP100A细胞中的表达。因此,外源过表达SP100A导致PML蛋白水平显著升高(图5M)。与此同时,IF分析显示了显著的PML小体在SP100A过表达细胞中的表达增加。总的来说,这些数据进一步证实了SP100A对PML的表达至关重要,与先前的研究一致。此外,ALKBH3缺陷细胞在PML小体中表现出显著的升高,这与ALKBH3在调节SP100A表达中的关键作用一致。综上所述,这些增益功能数据表明SP100A负责PML核凝聚物的形成,在眼部黑色素瘤中起肿瘤抑制作用。
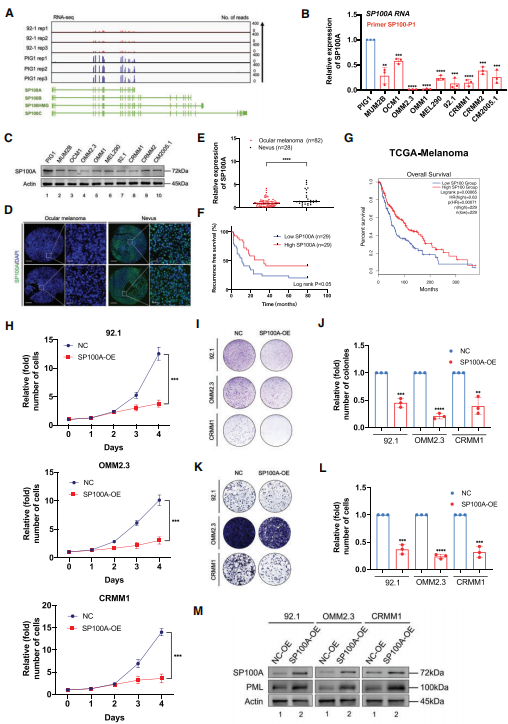
图5:SP100A在眼黑色素瘤中起抑瘤作用
(A) IGV从眼黑色素瘤细胞系(92.1)和正常黑色素细胞细胞系(PIG1)的RNA-seq数据中追踪SP100。
(B) qPCR数据显示,相对于PIG1细胞,SP100A在眼黑色素瘤细胞中的表达。
(C) Western blot显示眼黑色素瘤细胞和正常黑色素细胞中SP100A相对于ACTB的表达。
(D)肿瘤和正常标本中SP100A(绿色)和DAPI(蓝色)的免疫荧光。
(E)正常组织与肿瘤组织中SP100A水平的统计结果。
(F)低SP100A水平(n = 29)和高SP100A水平(n = 29)的眼部黑色素瘤患者肿瘤复发Kaplan-Meier曲线的差异。
(G)按SP100表达水平分层的TCGA黑色素瘤患者SP100表达与总生存率相关性的Kaplan-Meier分析:高(前50百分位,n = 229)和低(后50百分位,n = 229)。
(H) CCK8法检测SP100A过表达后眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)的增殖情况。
(I)采用集落形成实验评估SP100A过表达后眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)的生长情况。
(J) SP100A过表达后眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)集落形成实验数据的统计分析。
(K)采用transwell实验评估SP100A过表达后眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)的迁移情况。
(L) SP100A过表达后眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)的transwell检测数据统计分析。
(M) Western blot显示野生型和过表达SP100A的眼黑色素瘤细胞(92.1,OMM2.3和CRMM1)中PML相对于ACTB的表达。
(6)SP100A的沉默部分降低了alkbh3缺陷细胞的肿瘤抑制效果
为了进一步验证ALKBH3与SP100A表达之间的关系,我们通过转染已报道的针对SP100A的shRNA,进一步改变了ALKBH3抑制后眼黑色素瘤细胞中SP100A的表达。正如预期的那样,将SP100A- shRNA转染到三种眼部黑色素瘤细胞后,SP100A在RNA 和蛋白质水平上的表达均显著降低。在细胞生长中,SP100A的缺失部分恢复了由ALKBH3抑制引导的抑制(~ 50-60%),并且SP100A缺陷细胞对ALKBH3缺失的抵抗力更强(图6A)。同样,SP100A沉默的细胞比对照组有更多的菌落(图6B,红色和橙色列)。此外,抑制SP100A显著增强了细胞迁移能力,降低了ALKBH3缺陷黑色素瘤细胞的抑制效果(图6C)。最重要的是,敲低SP100A进一步挽救了ALKBH3-缺失的黑色素瘤细胞中原位肿瘤的形成(图6D-E)。与此一致,SP100A的稳定敲低导致野生型(图6F)和ALKBH3-缺失的眼部黑色素瘤细胞中PML的表达受损(图6F)。这些结果表明,ALKBH3在体内和体外均通过减少SP100A介导的PML小体来促进眼黑色素瘤。

图6:ALKBH3敲低的抗癌作用被SP100A沉默部分阻断
(A) CCK8试验评估了SP100A沉默后alkbh3缺陷眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)的增殖情况。
(B)通过集落形成试验评估SP100A沉默后alkbh3缺陷眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)的生长情况。
(C) transwell实验评估了在SP100A沉默后alkbh3缺陷眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)的迁移。
(D)体内小动物成像系统获得的图像显示,SP100A沉默后,来自ALKBH3缺陷92.1细胞的原位异种移植物的生物发光信号受到抑制。
(E) SP100A沉默后,来自ALKBH3缺陷92.1细胞的同种异种移植物的重量直方图。H&E染色显示肿瘤组织。
(F) Western blot显示ALKBH3和SP100A敲低后,眼部黑色素瘤细胞(92.1、OMM2.3和CRMM1)中ALKBH3、SP100A、PML相对于ACTB的表达。
(7)对SP100A进行m1A修饰可增强其RNA稳定性和翻译效能
然后,我们探索了SP100A的m1A修饰的表观遗传机制。由于已有研究表明YTHDF蛋白负责识别m1A甲基化,我们首先测试了SP100A mRNA与YTHDF1、YTHDF2和YTHDF3的结合状态。RNA免疫沉淀分析表明,YTHDF1特异性识别SP100A mRNA;然而,YTHDF2和YTHDF3仅表现出有限的相互作用强度(图7A)。此外,YTHDF1与SP100A在TCGA队列中的表达表现出显著的正相关(图7B),这与YTHDF1是识别SP100A所必需的假设完全一致。此外,YTHDF1的沉默显著抑制了SP100A的表达,并完全恢复了ALKBH3沉默介导的SP100A水平升高(图7C和D)。综上所述,这些数据表明YTHDF1是SP100A的读取蛋白。
然后,我们确定了SP100A mRNA的特异性m1A修饰位点。在SP100A的5UTR中,根据鉴定出的峰,我们发现了5个潜在的m1A位点c.44A (TGTGG),c.54A(AGACG)和c.67A(TGAGG)和 c.92A(图4I)。然后,我们将相应的野生型和突变的5UTRs克隆到pmirGLO载体中(图7E,上图)。荧光素酶报告基因分析表明 c.A92T的信号下降,而其他突变组的信号保持不变(图7E)。此外,我们采用RIP-qPCR来鉴定YTHDF1与报告基因转录本(F-luc)之间的相互作用频率。我们观察到pmirGLOMUT4 转录本在YTHDF1和F-Luc之间的结合亲和力降低,而其他转录本保持不变。这一结果与之前观察到的YTHDF1直接与m1A甲基化序列相互作用的结果一致,这被认为是RNA中m1A的“Reader”。此外,我们发现ALKBH3缺失的细胞中SP100A的RNA稳定性增强,这与ALKBH3缺失后SP100A RNA表达的增加相一致(图7F)。重要的是,在92.1细胞中进行的多聚体分析显示,ALKBH3的稳定敲除导致多聚体部分的SP100A mRNA丰度显著(图7G),这通常具有有效的翻译能力。这一观察结果与之前的结论一致,即早期外显子中的m1A修饰会导致其翻译能力的增强。值得注意的是,在ALKBH3基因敲低后,新生的SP100A RNA表达仍然没有发生改变。综上所述,这些结果表明,SP100A mRNA的m1A RNA甲基化有助于增加RNA的稳定性和转录后的翻译能力。

图7:对SP100A进行m1A修饰可提高其RNA稳定性和翻译效率
(A)通过YTHDF1、YTHDF2和YTHDF3检测SP100A在眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)中的表达。
(B) TCGA-UM队列中SP100A表达与YTHDF1表达的相关性分析(n = 80)。
(C) qPCR数据显示YTHDF1基因敲除后眼部黑色素瘤细胞(92.1、OMM2.3和CRMM1)中YTHDF1 RNA和SP100A RNA的表达。
(D) Western blot显示,ALKBH3和YTHDF1敲低后,眼黑色素瘤细胞(92.1、OMM2.3和CRMM1)中YTHDF1、SP100A、ALKBH3相对于ACTB的表达。
(E)荧光素酶报告基因测定显示了野生型和6个突变体SP100A5’UTR报告载体的相对荧光素酶活性。
(F)放xian菌素(5 g/ml)处理野生型和ALKBH3缺陷92.1细胞0-6小时SP100A的半衰期。
(G) ALKBH3敲除或不敲除92.1细胞的多体谱分析。提取不同核糖体组分的RNA,进行qPCR分析。
全文小结
本研究首次揭示了ALKBH3介导的m1A修饰在PML小体的形成中起抑制作用,为癌症中动态RNA修饰和相分离事件的理解架起了桥梁。
本研究发现SP100A的m1A修饰负责肿瘤抑制性PML小体的形成,这是第一个证明m1A是肿瘤抑制基因表达所必需的研究,扩展了目前对m1A动态功能的理解。
本研究发现组蛋白乳酸化也负责ALKBH3的致癌激活,导致癌症中的低m1A状态。
本研究首次发现SP100A在眼黑色素瘤中也具有肿瘤抑制作用,为视力和危及生命的眼黑色素瘤提供了新的治疗靶点。
总之,该研究结果揭示了一个全新的肿瘤发生模型,其中m1A修饰SP100A mRNA促进PML核凝聚物的形成。此外,本研究首次揭示了m1A参与肿瘤抑制基因的激活,揭示了组蛋白乳酸化、mRNA m1A修饰和相分离介导的凝聚物形成之间的串扰,从而提供了一种靶向m1A重编程的新型治疗方法,更有效地治疗肿瘤。
相关产品
m6A RNA甲基化测序
m5C RNA甲基化测序
m1A RNA甲基化测序
m7G RNA甲基化测序
ac4C RNA乙酰化测序
O8G RNA氧化修饰测序
2’-O-RNA甲基化测序
m6Am RNA甲基化测序
假尿嘧啶测序
RNA pulldown
RNA-seq
RIP测序
Ribo-seq
SLAM-seq
CHIRP-seq
CHIP-seq